June 4, 2026
30
min read
If your DHF has ever been questioned during an audit because an auditor could not follow a requirement from its source through to verification, you already understand the cost of weak traceability. This guide walks through exactly what a Requirements Traceability Matrix (RTM) must contain for medical device development, which regulatory clauses require it, where most teams fail, and how an integrated ALM+QMS platform eliminates the manual work of maintaining it.
The RTM is the documentary spine of your design control process. Without it, you cannot demonstrate that every user need has been translated into a design input, every design input has been implemented, and every implementation has been verified and validated.
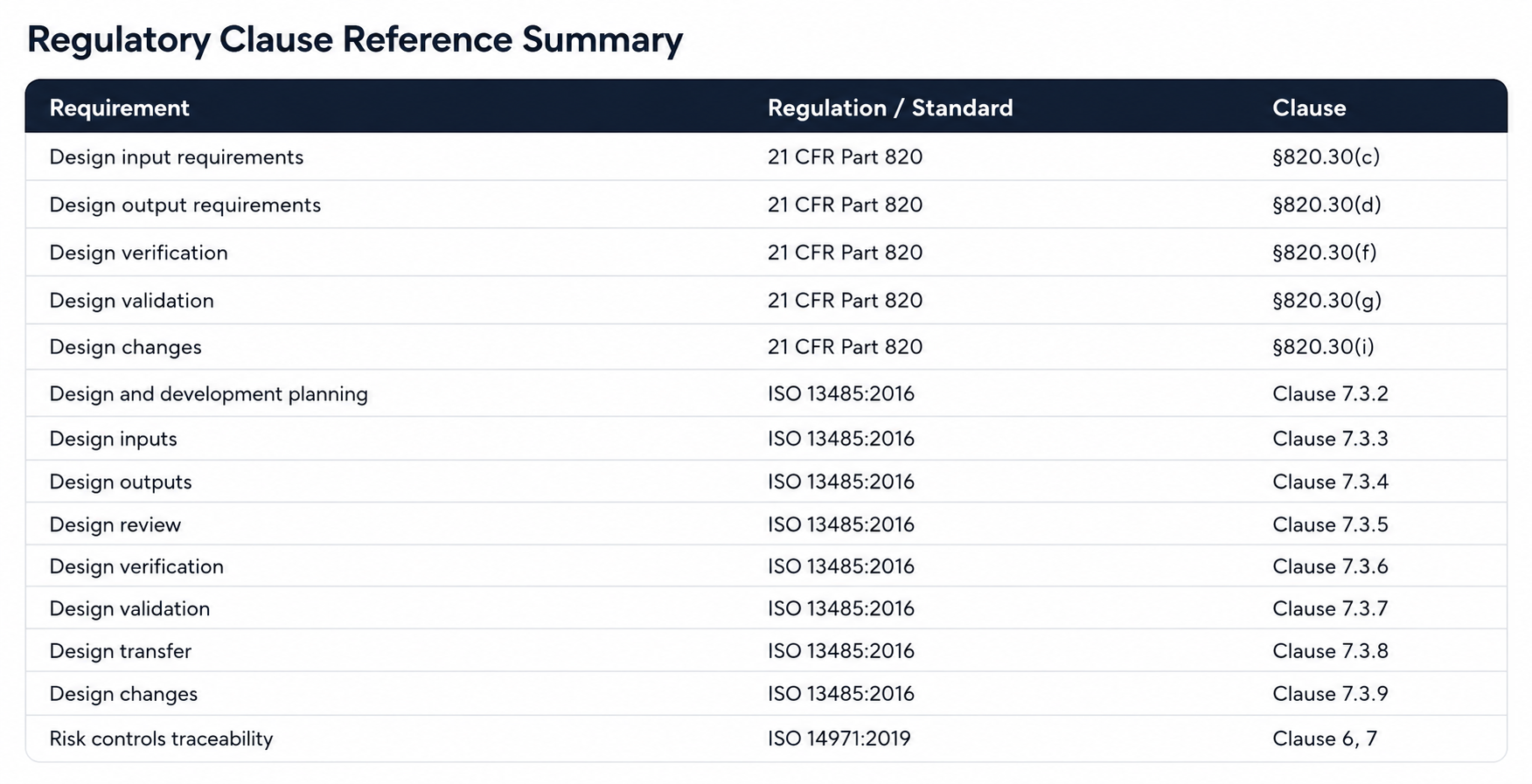
FDA 21 CFR Part 820.30 requires design controls that establish and maintain procedures to ensure device design is correctly translated into specifications. ISO 13485:2016 Clause 7.3 extends that requirement across the full design and development lifecycle, from planning through transfer. Neither regulation names the RTM explicitly, but both require the traceability it provides. In practice, every FDA investigator and ISO notified body auditor expects to see one.
The MDR (EU 2017/745) reinforces this through Annex II technical documentation requirements, which demand a description of the design stages applied and traceability between general safety and performance requirements and the evidence that satisfies them.
Failing to maintain an RTM is not a documentation style choice. It is a design control gap.
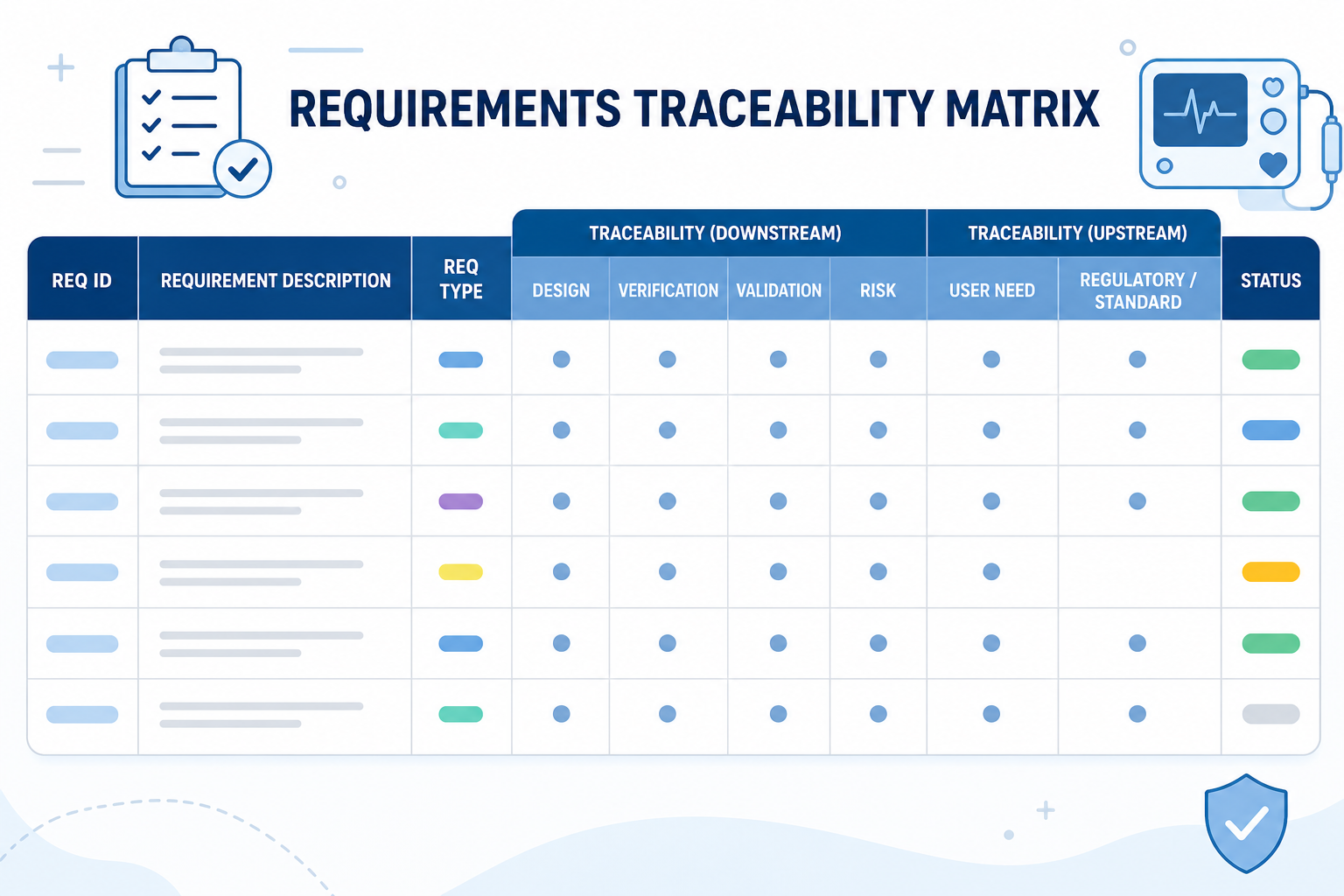
A minimal RTM maps requirements to tests. A compliant RTM for a medical device does significantly more. Here is what your matrix must trace:
Bidirectional traceability means you can move in both directions across the matrix:
An RTM that only maps forward is incomplete. Auditors will run both directions.
Many teams build an RTM that maps user needs to design inputs to test cases. They stop there. Validation records, post-market data links, and risk control evidence are kept in separate documents with no formal connection. Auditors see this as a broken chain.
The risk management file is maintained in a separate spreadsheet or document. The RTM has no column for risk controls. This means you cannot demonstrate at a glance that every hazard-related requirement has a corresponding control, and every control has been verified as effective.
Teams build the RTM late in development, populate it from memory, and treat it as a pre-submission artifact. When design changes happen (and they always do), the RTM is not updated. By the time an audit occurs, the matrix no longer reflects the actual device.
Without stable, unique requirement IDs, you cannot reliably link requirements across the matrix. "Requirement 7" in the design spec may or may not match "Requirement 7" in the test protocol. Auditors cannot reconcile them without interrogating your team.
No mechanism exists to identify orphaned requirements (inputs with no corresponding output or test) or orphaned tests (verification records that satisfy no stated requirement). These gaps are invisible until an auditor finds them.
For software-containing devices, IEC 62304 requires traceability from system requirements to software requirements to software architecture to unit tests. Teams often maintain this in a separate development tool with no linkage to the QMS-level RTM. The result: a fragmented traceability story across two systems.
A design change is processed through the CAPA or change control process, the design output is updated, but the RTM is not revised to reflect the impact. The matrix now shows a requirement verified against a version of the output that no longer exists.
Before building the matrix, define your requirement levels and naming convention. A typical hierarchy for a medical device:
Assign unique IDs at the point of creation. Do not renumber.
User needs belong in your design and development plan (ISO 13485:2016 Clause 7.3.2). They must be captured with enough specificity to be testable. "The device must be easy to use" is not a user need. "The device must allow a trained nurse to complete setup in under two minutes without reference to the instructions for use" is.
For every user need, identify the design inputs that satisfy it. A single user need may generate multiple design inputs. Document the rationale for the translation. This is the evidence that user needs were properly understood and interpreted.
For every design input with a safety or performance implication, check your risk management file. If a hazardous situation exists that the requirement addresses, create a formal link between the design input and the risk control. If no risk link exists, that absence must be a documented, justified decision.
Every design output must be traceable to at least one design input. If an output exists with no corresponding input, either a requirement is missing or the output should not exist. Run this check at every design review (ISO 13485:2016 Clause 7.3.5).
For each design input, identify the verification method (test, inspection, analysis, demonstration) and the corresponding protocol and report. For the full set of user needs, document how validation activities confirm the device meets intended use.
At each formal design review gate, baseline the RTM. Record which version of the RTM was reviewed and approved. This creates the audit trail showing that traceability was assessed and approved at each stage of development.
Every approved design change must include an RTM impact assessment as part of the change package. If a design input changes, the corresponding outputs, verifications, and risk links must be reviewed and updated.
Most medical device teams maintain their RTM across at least two systems: a requirements or ALM tool for engineering content, and a QMS platform for CAPA, design reviews, and DHF management. The gap between these systems is where compliance breaks down.
When a requirement changes in the ALM tool, someone must manually update the QMS. When a CAPA drives a design change, someone must trace its impact through the requirements. When an auditor asks for the full traceability chain from a user need through to a CAPA closed in response to a post-market complaint, you are assembling evidence across multiple disconnected databases.
This is not a process failure. It is a structural problem created by using tools that were not designed to work together.
Orcanos is built as a single platform that covers both ALM and QMS functions, which means the RTM is not a separate artifact you maintain alongside your quality records. It is a live view into the relationships that already exist across your development and quality data.
Within Orcanos, requirements are captured with unique IDs at the point of authoring. Design inputs link directly to user needs. Risk controls from the risk management module link to the same requirement records. Verification and validation tests are created and executed within the platform and automatically reflected in the traceability matrix. Design reviews reference the current state of the RTM, with electronic approval creating a timestamped, signed audit record.
When a design change is initiated, Orcanos surfaces every linked artifact: the requirements affected, the tests that need to be re-run, and the risk controls that need reassessment. The change cannot be closed without completing that impact chain. This is not a configuration add-on. It is how the system is structured.
For software-containing devices, Orcanos supports IEC 62304 traceability from system requirements through software requirements to test records, within the same platform that holds your ISO 13485 design controls and ISO 14971 risk records. The IEC 62304-to-ISO 13485 traceability gap that creates audit findings for so many teams is closed within a single system.
During an audit, the RTM in Orcanos is not assembled from exports. The auditor (or your QA team preparing for the audit) can navigate the live matrix, follow any link in either direction, and pull the underlying evidence without leaving the platform.
Use this during design reviews and pre-audit assessments:
If your current RTM lives in a spreadsheet or is split across an ALM tool and a separate QMS, the checklist above will surface gaps that are difficult to close without structural change.
Orcanos integrates your requirements, risk management, design controls, and CAPA into a single audit-ready platform, so your RTM is maintained automatically as your team works, not assembled manually before each audit.