April 26, 2026
8
min read
ISO 13485:2016 is the international standard for quality management systems in medical device manufacturing. Certification signals to regulators, notified bodies, and customers that your QMS is systematically designed to produce safe, effective devices. But certification is not the finish line. The standard demands ongoing operational discipline across design, production, risk management, and post-market surveillance.
This guide covers the core requirements, where companies typically break down, and how an integrated development and quality platform keeps your evidence audit-ready without manual assembly.
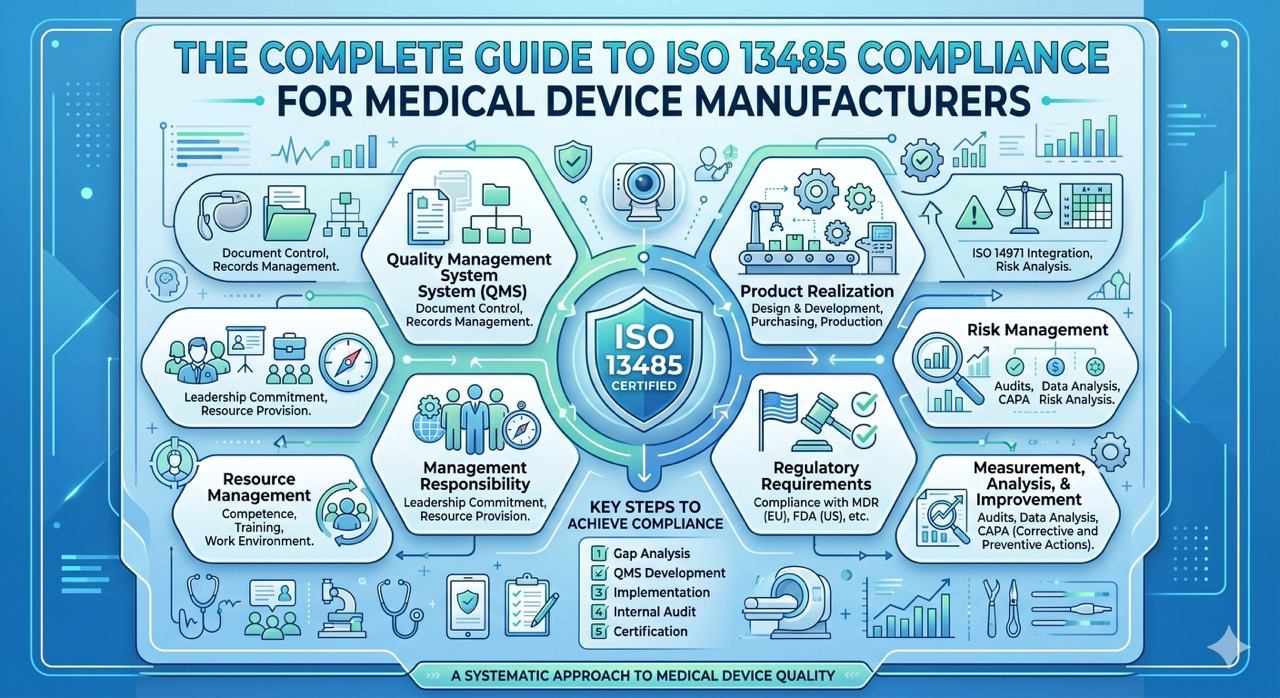
The standard is organized around eight sections. Sections 4 through 8 carry the substantive requirements.
Section 4: Quality Management System
You must document your QMS scope, maintain a quality manual (or equivalent documented structure), control your documents and records, and define how the system is reviewed and improved. Clause 4.2.3 governs document control; Clause 4.2.4 governs records control. Both require version history, approval workflows, and retention schedules tied to device lifetime.
Section 5: Management Responsibility
Top management must demonstrate active ownership of the QMS, not just sign a quality policy. This means documented management reviews (Clause 5.6) with inputs covering audit results, customer feedback, process performance, and CAPA status. The output must include resource decisions and improvement actions.
Section 6: Resource Management
This section covers personnel competency (Clause 6.2), infrastructure (Clause 6.3), and work environment (Clause 6.4). Auditors want to see training records tied to job roles and evidence that competency was verified, not just that training occurred.
Section 7: Product Realization
This is the densest section. It covers:
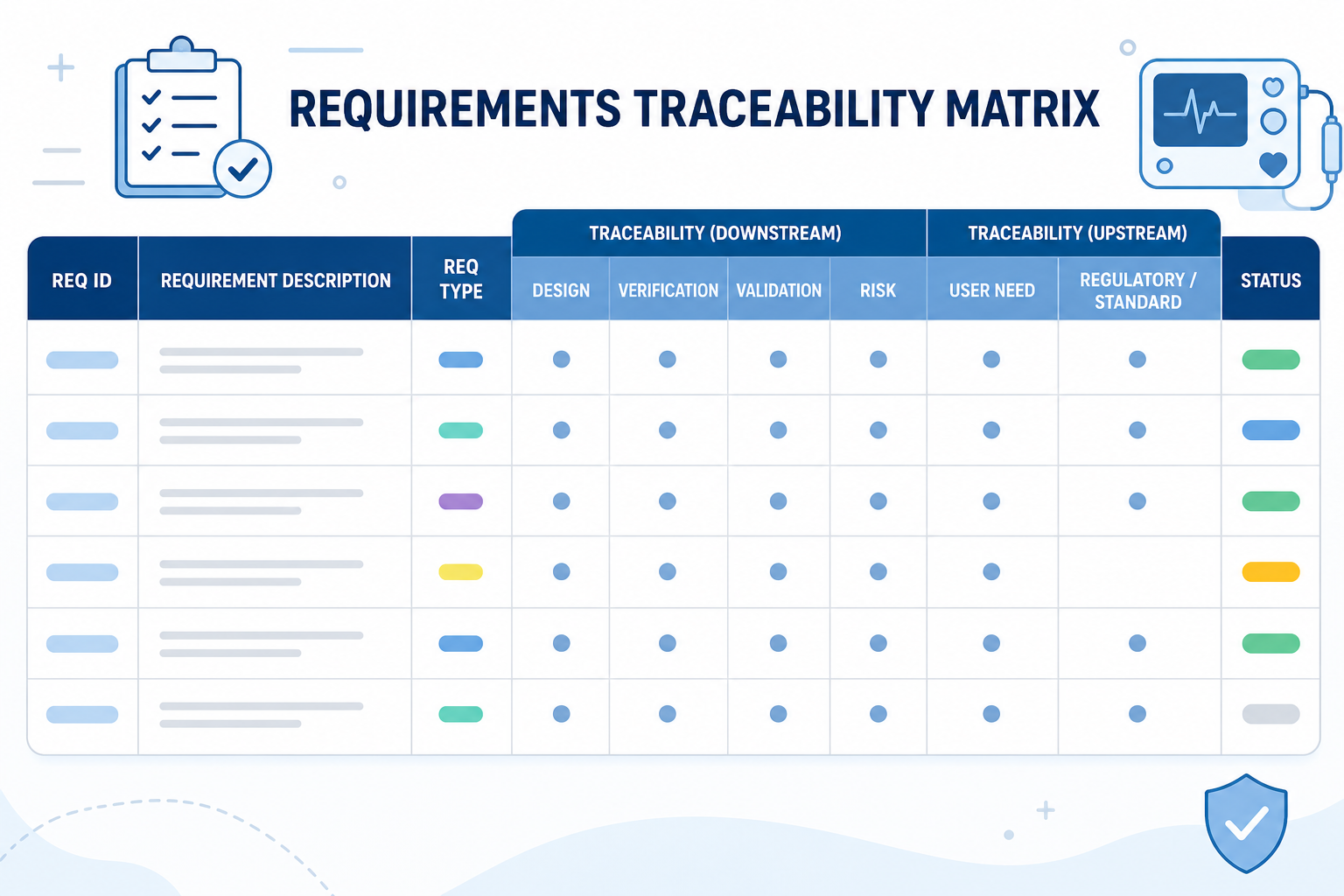
Design and development (Clause 7.3) is where most findings originate. It requires documented design inputs, outputs, reviews, verification, validation, and transfer. Critically, each stage must be traceable to the others. A design output must link back to the input it satisfies. A validation record must connect to the output it confirms.
Section 8: Measurement, Analysis, and Improvement
This section governs feedback loops: internal audits (Clause 8.2.2), monitoring and measurement of processes and product (Clauses 8.2.3 and 8.2.4), control of nonconforming product (Clause 8.3), data analysis (Clause 8.4), and improvement (Clause 8.5). CAPA lives here under Clause 8.5.2 (corrective action) and Clause 8.5.3 (preventive action).

1. Broken traceability in design controls
Clause 7.3.2 through 7.3.7 require a connected chain: user needs to design inputs, inputs to outputs, outputs to verification, outputs to validation. Most companies have these records. What they lack is a documented, navigable link between them. Auditors do not accept a folder of spreadsheets and a verbal explanation of how they connect.
2. Risk management isolated from design records
ISO 13485 requires integration with ISO 14971. Your risk management file must be maintained throughout the design lifecycle (Clause 7.1), not completed at the end. When risk controls appear in the design record but the risk file has no corresponding update, that is a finding.
3. CAPA without demonstrated effectiveness
Clause 8.5.2 requires corrective actions to be verified for effectiveness. Companies frequently document root cause analysis and the action taken, then close the CAPA. Without a documented effectiveness check linked to measurable criteria, the CAPA is non-conforming.
4. Supplier qualification records that cannot be retrieved
Clause 7.4.1 requires you to evaluate and select suppliers based on their ability to meet requirements. Auditors ask to see the evaluation records for your critical suppliers. If the records live in a filing cabinet, an inbox, or a spreadsheet with broken links, retrieval under audit conditions is a practical problem.
5. Document control failures during design changes
Clause 7.3.9 requires design changes to be identified, reviewed, verified, validated as appropriate, and approved before implementation. In practice, engineering makes a change, updates one document, and misses three others that reference the original specification. The review and approval record either does not exist or does not reference all affected documents.
Compliance is not a set of documents. It is a set of connected processes with evidence generated at each step. The operating model has four layers:
Layer 1: Document and record architecture
Every requirement in the standard maps to either a procedure (what you will do) or a record (proof that you did it). Build your document hierarchy intentionally: quality manual, procedures, work instructions, and forms. Clause 4.2.3 defines what document control must include. Apply it uniformly.
Layer 2: Design controls as the backbone of product records
The Device History File (DHF) is not created at submission. It accumulates throughout development. From the first design input to the final validation report, every record belongs in a structured design control framework. Version each document. Link each approval. Make the trace from user need to validated output navigable without interpretation.
Layer 3: Risk management woven into development, not appended to it
Under ISO 14971:2019 (referenced by ISO 13485 Clause 7.1), risk management activities run in parallel with design activities. Hazard identification informs design inputs. Risk controls become design outputs. Residual risk evaluation happens after verification. If your risk file and your design file are separate documents with no cross-references, the integration the standard requires is missing.
Layer 4: Continuous feedback through CAPA and audits
Complaints, nonconformances, audit findings, and trend data from post-market surveillance all feed into your CAPA system. Each CAPA record must document the source, the root cause method used, the action taken, and the effectiveness verification. The loop is not closed until effectiveness is confirmed.
The most common source of audit preparation pain is tool fragmentation. Requirements live in a requirements management tool. Risk records live in a spreadsheet. Design reviews are in email. CAPAs are in a separate QMS platform. When an auditor asks to see the connection between a design change and the risk record it affected, someone has to manually reconstruct that trail.
Orcanos addresses this directly by combining ALM and QMS functionality in a single platform. Requirements, design controls, risk records, CAPA, and the DHF all live in one environment with a shared data model. The traceability that ISO 13485 Clause 7.3 requires is built into the system structure, not assembled after the fact.
Specifically, this means:
For QA managers preparing for a Clause 7.3 audit deep-dive, this changes the preparation workload significantly. The evidence already exists in connected form. Retrieval is a query, not a project.
If your current compliance infrastructure involves more than two tools to answer a traceability question, the gap is structural, not procedural. Patching it with better spreadsheets does not resolve the audit risk.
Schedule a demo with Orcanos to see how connected ALM and QMS records work in practice, and how teams use the platform to walk auditors through a complete design control trace from user need to validated output without leaving the system.