May 12, 2019
13
min read

According to regulatory bodies, medical devices, pharmaceutical or automotive manufacturers should be implementing Quality Risk Management (QRM) systems when accessing the risks that come with the production of products that introduce safety issues to the user/operator/technician of the device. In this post, we will be addressing the relevant quality standards and regulatory guidelines.
Below are 5 crucial points from the FDA Guidance for Industry: Quality Systems Approach to mentioned industries cGMP regulations (2006).
21 CFR Part 11 – Scope and Application 2003: This guidance is the documentation of the FDA’s approach for electronic records and signatures. They recommend that for implementation of key requirements of Part 11 they should base the decision on a documented and justified risk assessment. Also, consideration should be given toward the potential of the system to influence product quality, record integrity, and safety.

Orcanos Electronic Signature
European Regulations: There is a legal status for Annex 15 to the EU GMPs Validation and Qualification. It utilizes a risk-based process for validation of changes to components, systems, facilities, and equipment. To determine the extent and scope of validation, use a Risk Assessment Approach. It crucial to always evaluate the possible effect of changes in facilities, equipment, and systems on the product. Also, do not forget to include a risk analysis report.
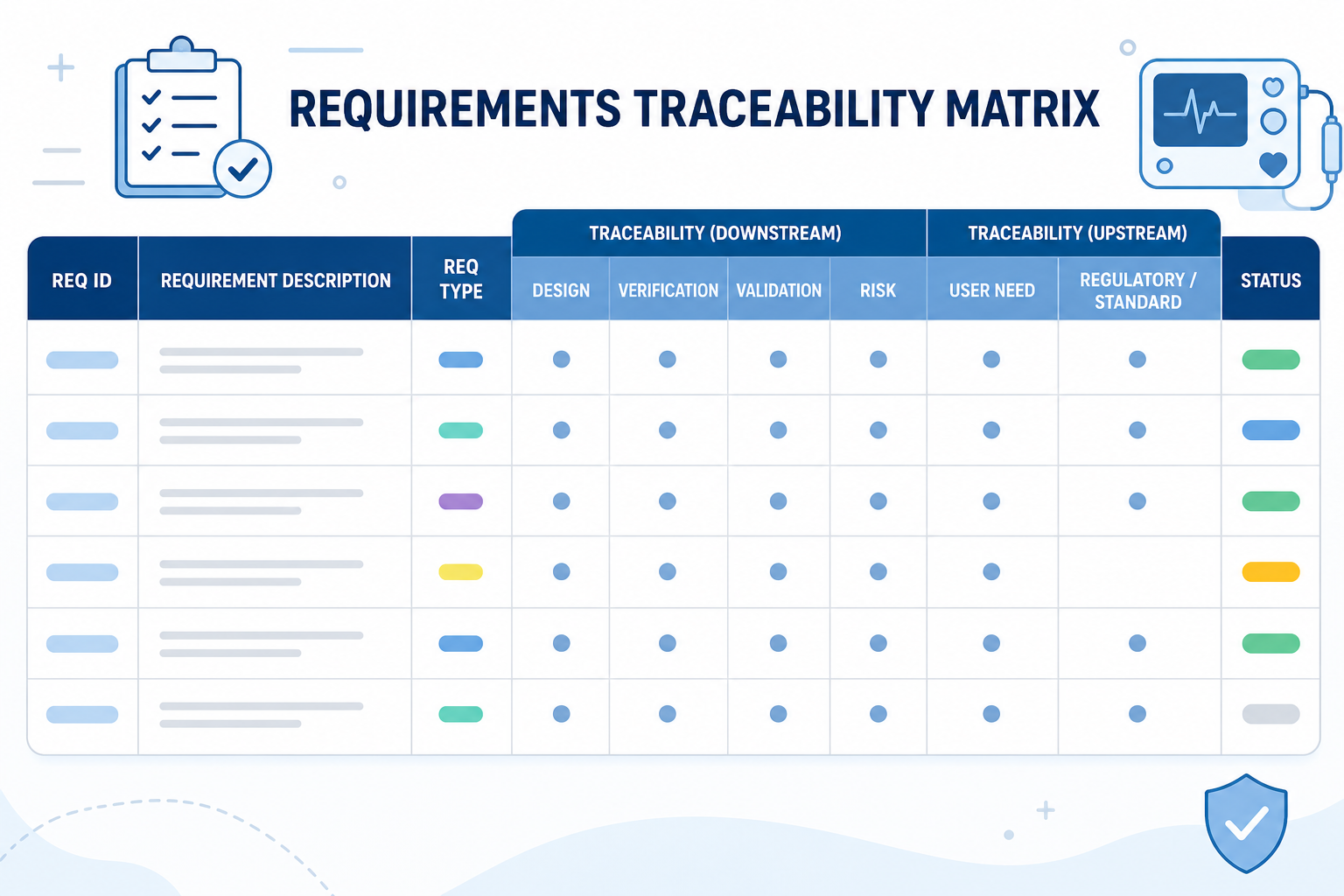
In the Orcanos Design Control module, there are several tools that allow you to conduct the assessment of a change. It gives you tools to raise subspecies indicators which are based on existing traceability between artifacts or to asses risk to newly introduce change as part of the risk assessment process.

Orcanos Suspicious Effect on Change Report
There is a legal status for Annex 11 to the EU GMPs for using computerized systems. It mandates that control for computerized systems be based on a document and justified risk assessment. Likewise, the level of validation and data controls have to be in line with a proper and documented risk assessment.
Orcanos computerized system is a validated system and it comes with a validation package that is complying with the regulatory requirements for both the USA and EU.
International Conference for Harmonisation ICH Q9/ISO 14971/ISO 26262: It is regarded as the most important document when it comes to risk assessment for the pharmaceutical/medical device/automotive correlated sector. The ICH focuses on how scientific knowledge plays a role in the protection of patients in the life industry and ISO 14971 focuses on the hazardous situation and potential mitigation while the ISO 26262 focus on safety issues related to critical components. These reference documents provides guidance on implementation processes. The three major principles that govern quality risk management are;
It might not always be necessary or appropriate to make use of a formal risk management process. For instance, using a well-known tool for an internal process like a standard operating procedure. Similarly, it is acceptable to use informal risk management processes or standard operating procedures for internal processes.
International Standards Organisation (ISO): The organization has three standards that address risk management. They are;
NOTE: There is a major difference between ISO 31000 and ISO 31010 despite addressing the same risk management problems. ISO 31000 highlights principles and guidelines, while ISO31010 describes risk assessment techniques.
ISO 14971: 2012 (Application of Risk Management to Medical Devices): While this document is for medical devices, the FDA recommends it to the pharmaceutical industry, others recommended it for the automotive as well. In this international standard, there is an outline that manufacturers can follow to identify potential hazards that concern medical devices. Also, it covers in-vitro diagnostic devices (IVD). Using the stipulated process, manufacturers can evaluate, and estimate the level of risk and take actions to control and monitor the risks. Likewise, the manufacturers will have criteria that will help them determine the effectiveness of the controls.
Throughout the lifecycle of any medical device, the requirements in this standard are applicable. However, it does not extend to clinical decision making, neither does it specify what risks levels are acceptable. Finally, the standard does not mandate that manufacturers use a quality management system. Nevertheless, a risk management system is a crucial part of the quality management system.
Reference Links