Products
ALM
Application Lifecycle Management
QMS
Quality Management System
Document Control
Document Management System
Features
Risk Management
Requirements Management
Test Management
Traceability Management
Defect Tracking
Customer Service
Ask Paul - AI Assitant
Complaints Management
CAPA Management
Engineering Change Order
Audit Management
Supplier Qualification
Training Management
Resources
Blog
Orcanos Academy
Events & Webinars
Help Center
Security Center
What's New
Company
About Us
Partners Program
Contact Us
Customers
Pricing
Get started
Get a Demo
Orcanos Blog
March 4, 2026
10
min read
When Your QMS Vendor is Acquired by its competitor: A Survival Guide for MedTech
Zohar Peretz
Co-Founder & Business Development
Compliance
October 29, 2024
4
min read
Summus Laser Selects Orcanos to Revolutionize Quality Management in the Laser Therapy Market
News
March 5, 2024
8
min read
Best QMS Software for Medical Devices: A Review of Orcanos
News
October 11, 2023
11
min read
Empowering Your Quality Management with AI-Driven Automation
Best Practices
Latest Articles
Compliance
Best Practices
Tips
News
Technology
June 4, 2026
30
min read
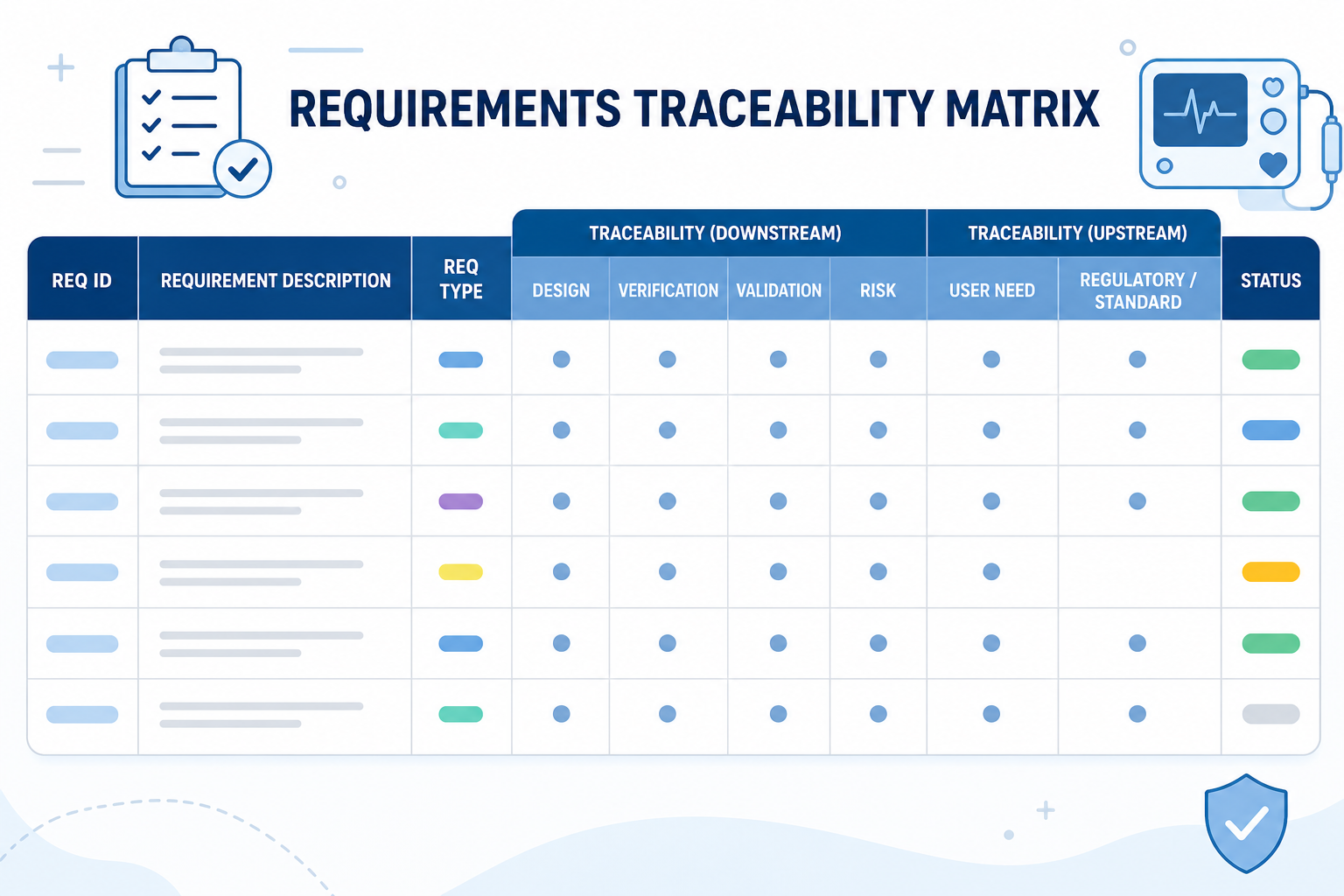
Requirements Traceability Matrix for Medical Devices: A Complete Compliance Guide
Compliance
May 18, 2026
19
min read
The Best Quality Management System for Medical Device Companies: An Honest Evaluation
News
April 26, 2026
8
min read
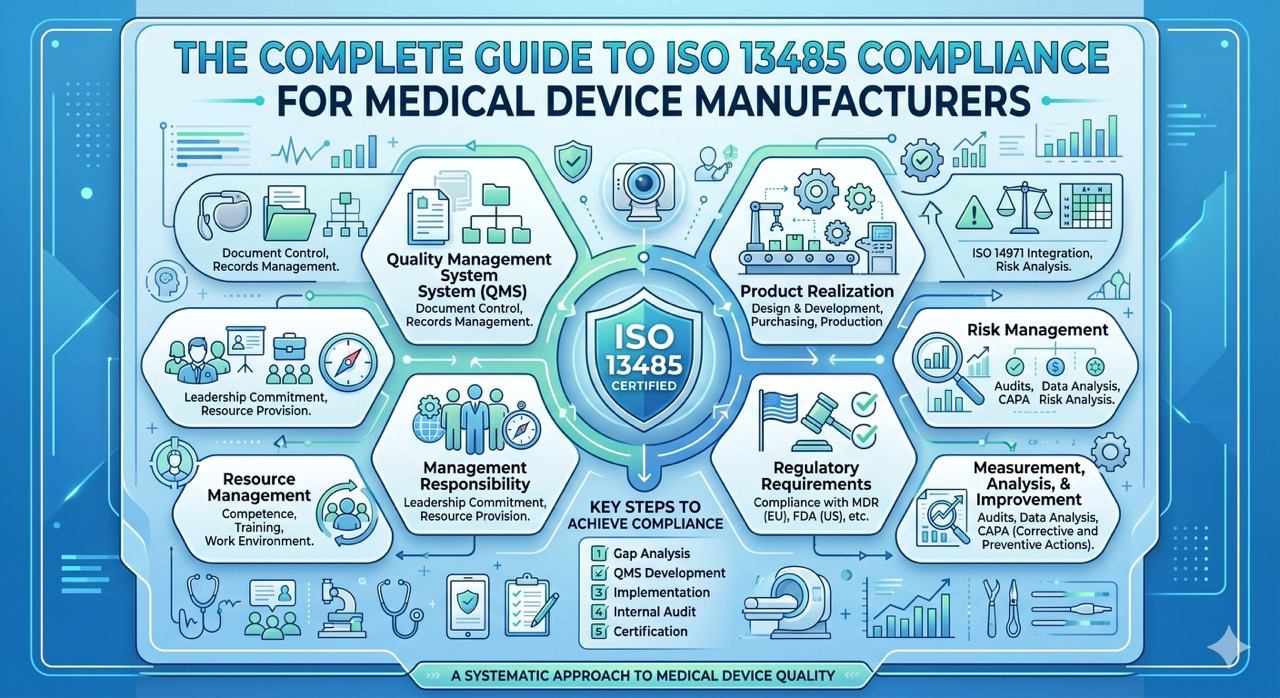
The Complete Guide to ISO 13485 Compliance for Medical Device Manufacturers
Compliance
April 15, 2026
5
min read
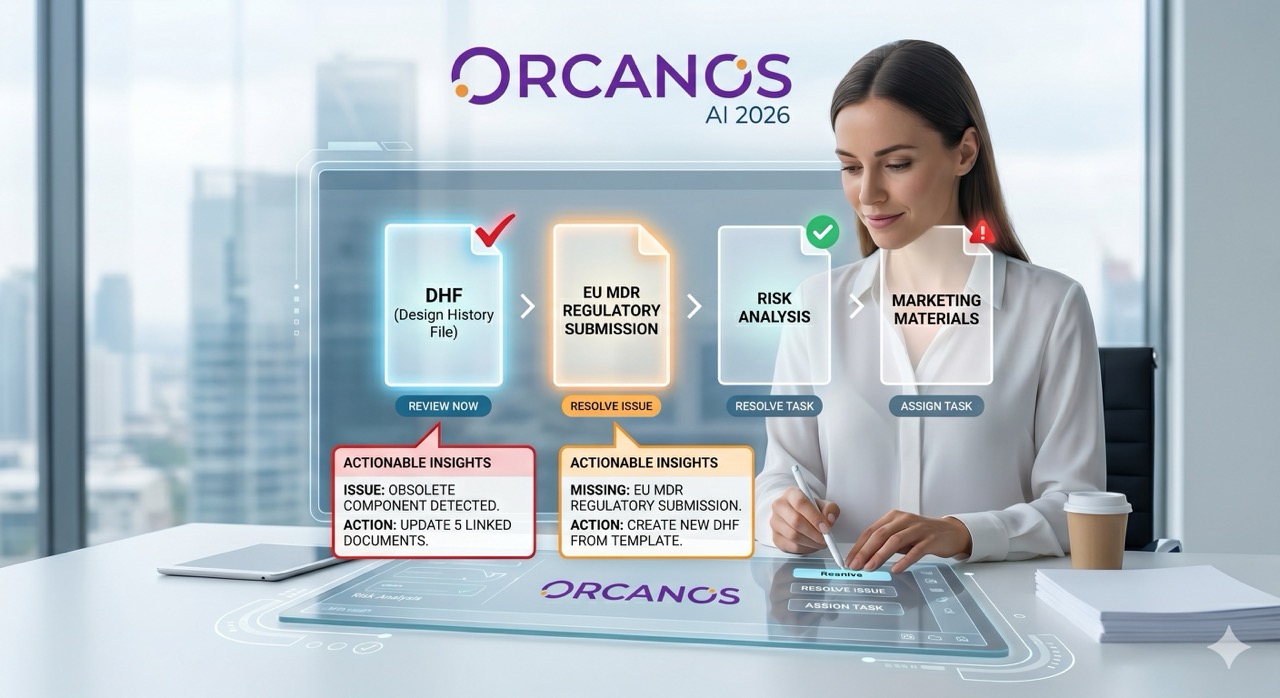
Orcanos AI: Intelligent Document Analysis That Transforms Regulatory Compliance
News
March 9, 2026
7
min read
Stop Fearing the Knock: Master Your Next Medical Device Audit
Compliance
March 4, 2026
10
min read
When Your QMS Vendor is Acquired by its competitor: A Survival Guide for MedTech
Compliance
July 27, 2025
8
min read
Simplifying Traceability in Orcanos
Compliance
May 29, 2025
min read
Choosing the Right Requirements Management Software in Regulated Industries
Best Practices
May 22, 2025
14
min read
CAPA in the Pharmaceutical Industry: From Compliance Burden to Strategic Advantage
Next
Trusted by